
Durante la pandemia de Covid, el gobierno de EE. UU. gastó miles de millones de dólares en casi 400 productos destinados a proteger, diagnosticar y tratar a cientos de millones de personas, todos con la etiqueta “EUA” o “Autorización de uso de emergencia”.
Pero ¿qué significa realmente EUA?
Incluso antes de responder a esa pregunta, y para entender dónde se encuentra la EUA en relación con otras vías para autorizar o aprobar productos médicos, es útil analizar qué EUA no es:
EUA no es una designación para un producto experimental que se somete a un ensayo clínico
Si solo entendiéramos una cosa sobre la EUA, debería ser esta: la EUA no se aplica a un producto sometido a un ensayo clínico regido por las regulaciones de la FDA u otros requisitos legales.
EUA tampoco es lo mismo que Uso de Acceso Ampliado (EAU), a menudo llamado acceso de “uso compasivo”, que se aplica para otorgar a los pacientes con enfermedades graves e incurables acceso a productos experimentales antes de que sean aprobados por completo.
Esta tabla de un Presentación FDA-CDC 2020 resume las diferencias entre los productos sometidos a ensayos clínicos, los productos administrados a los pacientes a través de un acceso "compasivo" ampliado y los productos autorizados a través de EUA:
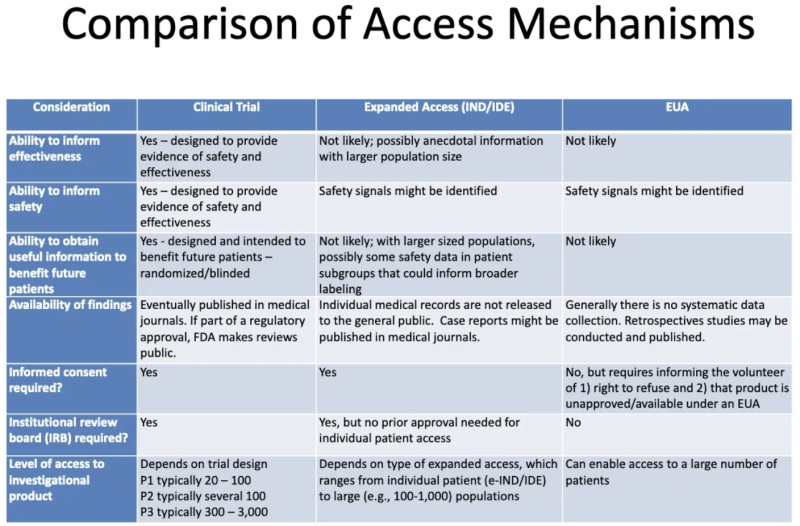
Esto es lo que nos dice esta tabla sobre EUA:
- No es probable que el proceso de concesión de la EUA genere información sobre la eficacia de un producto.
- El proceso de concesión de EUA no está diseñado para proporcionar evidencia de seguridad o eficacia, pero se pueden identificar señales de seguridad.
- Es poco probable que, una vez que se concede la EUA a un producto y se administra a algunos pacientes, se obtenga información útil que beneficie a futuros pacientes.
- No existe una recopilación sistemática de datos sobre la eficacia o seguridad de la EUA, y no se publican datos en revistas médicas como parte del proceso de aprobación regulatoria.
- No se requiere consentimiento informado, pero a los pacientes que se “ofrecen voluntariamente” a tomar el producto se les debe informar que pueden negarse y que el producto no está aprobado/disponible según la EUA.
- No se requiere una junta de revisión institucional (IRB). [IRB es una junta que se supone protege el bienestar de sujetos humanos en ensayos clínicos]
Para aclarar aún más cuán separada está la EUA de cualquier proceso de aprobación normal, en un 2009 Publicación del Instituto de Medicina de las Academias Nacionales, encontramos esta declaración:
Es importante reconocer que una EUA no es parte del camino del desarrollo; es una entidad completamente separada que se utiliza sólo durante situaciones de emergencia y no forma parte del proceso de aprobación de medicamentos. (pág. 28)
En resumen:
Es poco probable que el proceso de concesión de una EUA a un producto genere evidencia de seguridad o eficacia. Una vez que se concede la EUA a un producto y se administra a los pacientes, es poco probable que se obtenga información útil que beneficie a futuros pacientes, porque no existe una recopilación sistemática de datos sobre su eficacia o seguridad.
Con base en toda esta información muy clara de los CDC/FDA y la IMNA, sería justo concluir que la Autorización de Uso de Emergencia es un proceso que debe aplicarse con mucha prudencia y sólo en casos de emergencias extremas.
Ahora veamos para qué tipos de situaciones de emergencia está diseñada legalmente la EUA.
La EAU está destinada a emergencias por armas de destrucción masiva
Las leyes que permiten el “Mecanismo de Acceso” de la EUA descrito anteriormente fueron redactadas para casos de emergencias extremas e inmediatas que involucran armas de destrucción masiva (ADM), también conocidas como agentes QBRN (químicos, biológicos, radiológicos y nucleares).
Así es como la Administración de Alimentos y Medicamentos (FDA) describe sus poderes de EUA:
Sección 564 de la Ley FD&C (21 USC 360bbb – 3) permite a la FDA fortalecer la protección de la salud pública contra agentes biológicos, químicos, nucleares y radiológicos.
Con esta autoridad de EUA, la FDA puede ayudar a garantizar que se puedan utilizar contramedidas médicas en emergencias para diagnosticar, tratar o prevenir enfermedades o afecciones graves o potencialmente mortales causadas por agentes biológicos, químicos, nucleares o radiológicos cuando no existen medidas adecuadas y aprobadas. y alternativas disponibles (entre otros criterios).
Estos poderes de EUA se otorgaron en 2004 en circunstancias muy específicas relacionadas con la preparación para ataques de agentes QBRN.
Como se explica en la Ley de Salud de Harvard Law,
En última instancia, fue la Guerra contra el Terrorismo la que daría lugar a la autorización de uso de emergencia. Después de los acontecimientos del 11 de septiembre de 2001 y los posteriores ataques por correo con ántrax, el Congreso promulgó la Ley del Proyecto Bioshield de 2004.
El grabar indica que el Congreso se centró específicamente en la amenaza del bioterrorismo, no en prepararse para una pandemia de origen natural.
Dado un tipo tan limitado de situación de emergencia verdaderamente extrema que involucra un ataque con armas de destrucción masiva, es comprensible por qué el “mecanismo de acceso” de la EUA no requiere mucha supervisión regulatoria ni cumplimiento de ningún estándar de fabricación o ensayo clínico.
Entonces, ¿qué requiere realmente el mecanismo de acceso a la EUA?
Los 3 pasos para la autorización de uso de emergencia (EUA)
Tienen que suceder tres cosas para que se conceda la EUA a un producto médico:
- El Secretario de Seguridad Nacional, el Secretario de Defensa o el Secretario de Salud y Servicios Humanos deben determinar que existe una emergencia que involucra un ataque o una amenaza de ataque con un agente QBRN o una enfermedad causada por dicho agente.
- La FDA debe asegurarse de cumplir con cuatro “criterios legales” cuando emite la EUA.
- La FDA tiene que "imponer ciertas condiciones requeridas" en la EUA.
Paso 1 de la EUA: Declarar una emergencia QBRN
La declaración de emergencia para EUA es independiente y no está relacionada con ninguna otra declaración de emergencia que pueda emitir el presidente, el secretario del HHS o cualquier otra persona. Debe emitirse específicamente con el fin de activar la EUA y puede finalizarse o ampliarse independientemente de cualquier otra declaración de emergencia.
Esto es lo que la ley EUA establece Son los cuatro escenarios posibles para activar el “mecanismo de acceso” de la EUA:
- una determinación del Secretario de Seguridad Nacional de que existe una emergencia interna, o un potencial significativo para una emergencia interna, que implica un mayor riesgo de ataque con un agente o agentes biológicos, químicos, radiológicos o nucleares;
- una determinación por parte del Secretario de Defensa de que existe una emergencia militar, o un potencial significativo para una emergencia militar, que implica un mayor riesgo para United Zonas fuerzas militares, incluido el personal que opera bajo la autoridad del Título 10 o el Título 50, de ataque con:
- un agente o agentes biológicos, químicos, radiológicos o nucleares; o
- un agente o agentes que puedan causar, o estén asociados de otro modo con, un riesgo específico y que ponga en peligro la vida de United Zonas fuerzas militares;
- una determinación por parte del Secretario [de Salud y Servicios Humanos] que existe una emergencia de salud pública, o un potencial significativo para una emergencia de salud pública, que afecta, o tiene un potencial significativo de afectar, la seguridad nacional o la salud y seguridad de United Zonas ciudadanos residentes en el extranjero, y que involucre un agente o agentes biológicos, químicos, radiológicos o nucleares, o una enfermedad o condición que pueda ser atribuible a dicho agente o agentes; o
- la identificación de una amenaza importante de conformidad con la sección 319F-2 de la Ley de servicios de salud pública [42 USC 247d-6b] suficiente para afectar la seguridad nacional o la salud y seguridad de United Zonas ciudadanos residentes en el extranjero.
Paso 2 de la EUA. Cumplimiento de los criterios legales
Una vez que uno de los secretarios ha declarado que existe una emergencia que justifica la EUA, hay cuatro “criterios estatutarios” más que deben cumplirse para que la FDA emita la EUA. Así es como la FDA explica estos requisitos:
- Enfermedad o afección grave o potencialmente mortal
Para que la FDA emita una EUA, los agentes QBRN mencionados en la declaración EUA del Secretario del HHS deben ser capaces de causar una enfermedad o afección grave o potencialmente mortal.
- Evidencia de efectividad
Los productos médicos que pueden considerarse para una EUA son aquellos que “pueden ser efectivos” para prevenir, diagnosticar o tratar enfermedades o afecciones graves o potencialmente mortales que pueden ser causadas por uno o varios agentes QBRN identificados en la declaración de autorización del Secretario del HHS. emergencia o amenaza de emergencia bajo la sección 564(b).
El estándar de “puede ser eficaz” para las EUA proporciona un nivel de evidencia más bajo que el estándar de “eficacia” que utiliza la FDA para la aprobación de productos. La FDA tiene la intención de evaluar la eficacia potencial de un posible producto EUA caso por caso mediante un análisis de riesgo-beneficio, como se explica a continuación.
[NEGRITA AÑADIDA]
- Análisis de riesgo / beneficio
Un producto puede ser considerado para una EUA si el Comisionado determina que los beneficios conocidos y potenciales del producto, cuando se usa para diagnosticar, prevenir o tratar la enfermedad o condición identificada, superan los riesgos conocidos y potenciales del producto.
Para determinar si los beneficios conocidos y potenciales del producto superan los riesgos conocidos y potenciales, la FDA tiene la intención de mirar la totalidad de la evidencia científica para realizar una determinación global riesgo-beneficio. Tal evidencia, que podría surgir de una variedad de fuentes, puede incluir (pero no se limita a): resultados de ensayos clínicos nacionales y extranjeros, datos de eficacia in vivo de modelos animales y datos in vitro, disponible para consideración de la FDA. La FDA también evaluará la calidad y cantidad del evidencia disponible, dado el estado actual del conocimiento científico.
[NEGRITA AÑADIDA]
- Sin alternativas
Para que la FDA emita una EUA, no debe haber ninguna alternativa adecuada, aprobada y disponible al producto candidato para diagnosticar, prevenir o tratar la enfermedad o afección. Un posible producto alternativo puede considerarse “no disponible” si no hay suficientes suministros de la alternativa aprobada para satisfacer plenamente la necesidad de emergencia.
Paso 3 de la EUA. Imponer las condiciones requeridas
Una vez que tengamos la declaración de emergencia específica de la EUA, y una vez que la FDA determine que el producto puede ser efectivo y que cualquier evidencia disponible demuestre que sus beneficios superan sus riesgos, hay una capa más de regulación relacionada.
Así es como un Informe del Servicio de Investigación del Congreso de 2018 sobre EUA explica esto:
FFDCA §564 ordena a la FDA que imponga ciertas condiciones requeridas en una EUA y permite condiciones discrecionales adicionales cuando corresponda. Las condiciones requeridas varían dependiendo de si la EUA es para un producto no aprobado o para un uso no aprobado de un producto aprobado. Para un producto no aprobado, las condiciones de uso deben:
(1) garantizar que los profesionales de atención médica que administran el producto reciban la información requerida;
(2) garantizar que las personas a quienes se administra el producto reciban la información requerida;
(3) prever el seguimiento y notificación de eventos adversos asociados con el producto; y
(4) prever el mantenimiento de registros y la presentación de informes por parte del fabricante.
Conclusión
Como se señala en este artículo, la FDA y los CDC reconocen claramente que es poco probable que el proceso de concesión de Autorización de uso de emergencia (EUA) genere información sobre la eficacia o seguridad de un producto. Cuando miramos la letra de la ley que rige la EUA, vemos que ésta es, de hecho, una evaluación correcta.
La ley EUA no impone ningún estándar legal o regulatorio que pueda determinar si un producto es seguro o efectivo. Los únicos estándares son si la FDA cree que el producto puede ser efectivo y que sus beneficios conocidos superan sus daños conocidos. Si no se conocen daños ni beneficios, porque el producto nunca ha pasado por el proceso de aprobación de medicamentos, la FDA puede utilizar cualquier información o estándar que elija para tomar esa determinación.
De todo esto se deduce que una empresa cuyo producto es candidato a EUA puede intentar demostrar la seguridad y/o eficacia del producto a través de cualquier medio que elija. La existencia de tal intento (ya sea un ensayo clínico u otro mecanismo de recopilación de datos) y cómo se lleva a cabo ese intento depende de la empresa. Nada en la ley EUA se aplica a cómo la empresa diseña, realiza o analiza cualquier estudio u otro mecanismo de recopilación de datos que decida realizar.
Aplicado a los productos Covid esto significa:
- No se requirieron datos de seguridad o eficacia de ensayos clínicos para que los productos Covid recibieran EUA.
- Todos los ensayos clínicos a los que se hace referencia en el proceso de EUA se llevaron a cabo sin estándares regulatorios legalmente aplicables.
- Cuando descubrimos que estos productos carecen de eficacia o seguridad, no es una sorpresa. Es un resultado muy probable del proceso.
- No hay datos del proceso EUA en los que basar decisiones fuera de la EUA sobre la seguridad o eficacia del producto. Por lo tanto, cualquier uso del producto fuera de la EUA requeriría pasar por el proceso de aprobación legal para productos médicos regulares desde el principio.
Más sobre el proceso de aprobación de las vacunas Covid esta página.
Reeditado del autor Substack
Publicado bajo un Licencia de Creative Commons Atribución Internacional
Para reimpresiones, vuelva a establecer el enlace canónico en el original Instituto Brownstone Artículo y Autor.








