

Durante las últimas décadas de mi carrera, he pasado innumerables horas trabajando para proteger a los estadounidenses investigando la seguridad de los medicamentos. Mi educación y mi carrera me han llevado a través de media docena de universidades, las grandes farmacéuticas y la FDA durante tres administraciones presidenciales. La seguridad de los medicamentos considera por qué un individuo puede tomar un producto farmacéutico y no tener ningún evento adverso, mientras que un individuo diferente podría tomar el mismo producto pero tener reacciones adversas que pueden llegar hasta la discapacidad permanente o la muerte. Por defecto, el estudio de la seguridad de los medicamentos también considera aspectos no clínicos de la fabricación y la calidad de los medicamentos.
Debido a que la calidad de los medicamentos es un factor esencial en la evaluación de la seguridad de los medicamentos, mi viaje para proteger a los estadounidenses me llevó a conceptualizar y fundar la primera "farmacia analítica” tiene la misión de verificar científicamente productos farmacéuticos de lugares como India y China antes de dispensarlos a los pacientes. Desafortunadamente, la búsqueda de generosidad por encima de la ética y la protección de los pacientes llevó a que la dirección financiera de esa empresa se comprometiera en los detalles Violaciones de la FDA y ser acusado por los jueces de hacer afirmaciones científicas falsas (Todo lo cual ocurrió accidentalmente después de mi salida).
Sin una confirmación externa de la calidad de los medicamentos, los estadounidenses dependen completamente de la FDA y de los fabricantes para evaluar y confirmar la pureza del producto. Se ha demostrado que la seguridad de los medicamentos es un problema notable cuando se trata de inyecciones de ARNm de Covid. Desafortunadamente, si alguien quisiera realizar su propio análisis de inyecciones de ARNm, tendría que hacerlo. no tienen una lista de ingredientes adecuadamente detallada con la que compararlos, ni siquiera acceso a la metodología regulatoria establecida sobre cómo probar adecuadamente su pureza.
Es porque los fabricantes y La FDA considera todos los ingredientes de estas inyecciones de ARNm, incluida la secuencia de ARNm más las propiedades de las nanopartículas lipídicas (LNP), incluida la vida media, las estructuras de LNP, las modificaciones de la superficie, el número/tipo(s) de LNP por dosis y los puntos de unión en la cadena de ARNm, no especificada o "secreto comercial".
Además de eso, la FDA también considera la metodologías sobre cómo probar la pureza de las inyecciones de ARNm también es un secreto comercial.
Apoyo bipartidista y cientos de miles de millones de dólares de los contribuyentes, pero ¿NO hay transparencia?
El secreto del ARNm de Covid existe a pesar de que las administraciones de Trump y Biden habían propuesto una transparencia total con las inyecciones de ARNm hasta el punto de eliminar los derechos de propiedad intelectual del ARNm de Covid. A pesar de eso, tanto la FDA como los fabricantes permiten o mantienen un estricto control de las patentes, incluidos los datos básicos sobre estas inyecciones, como secreto comercial. Lo hacen a pesar de que todos los fabricantes de vacunas Covid han recibido cientos de millones de dólares de los contribuyentes según Forbes/Statista publicaciones.
Estudiar la epidemiología de la seguridad de los medicamentos ya es bastante difícil. Sin una pureza/consistencia verificable del producto, es imposible realizar una evaluación de seguridad completa.
La transparencia total de todos los ingredientes y las medidas de control de calidad son importantes no solo porque fueron financiadas en gran medida por los contribuyentes con cientos de millones de dólares, sino porque han surgido una serie de preguntas sobre la seguridad y eficacia de las inyecciones de ARNm de Covid.
Además de ser excepcionalmente compleja, su aprobación fue acelerada por los reguladores después menos de un año. La mayoría de los medicamentos y vacunas suelen tardar alrededor diez años para probar completamente la seguridad/eficacia y revisar y aprobar. Además de que los ingredientes son completamente novedosos, muy complejos y los primeros de su tipo en administrarse a escala masiva, el desarrollo incluye Las evaluaciones clínicas de seguridad/toxicidad a largo plazo y las revisiones epidemiológicas se aceleraron y probablemente no se aclararon completamente antes de su lanzamiento.
La verificación de ingredientes, la transparencia y la “veracidad” de la FDA tienen precedentes que se remontan al siglo XIX:
La verificación analítica y la transparencia de ingredientes o “verdad en el etiquetado” donde se indica el contenido de la botella Requisitos para que coincida con los ingredientes enumerados Es anterior al establecimiento de la FDA, se remonta a 1862.. La FDA actual en realidad nació de lo que comenzó como un único empleado del “Departamento de Química” empleado en el Departamento de Agricultura de Estados Unidos.
Adulteración, (ingredientes alterados o tóxicos) malinterpretar (contiene una etiqueta falsa o es engañoso o contiene afirmaciones médicas incorrectas), o etiquetado incorrecto (contiene ingredientes que no figuran en la etiqueta del producto) han tenido historias largas y desagradables en Estados Unidos. Se pensaba que la atrocidad había alcanzado su punto máximo entre principios y mediados del siglo XIX (o al menos fue entonces cuando se volvió identificable), ya que recién en 19 se habían desarrollado procesos técnicos para analizar y detectar el fraude de ingredientes. Antes de eso, los llamados “curanderos ambulantes” que se hacían llamar “médicos” (invariablemente con credenciales dudosas o inexistentes) vendían frascos de productos que “lo curan todo”, cuyas etiquetas de ingredientes sólo enumeraban contenidos nebulosos o inocuos, como “vitaminas""extractos de hierbas,"O"aceite de serpiente” – o, a menudo, no tienen ninguna lista de ingredientes.
En aquel entonces, muchos habitantes de Nueva Inglaterra devotos y puritanos, que por razones religiosas preferirían nunca tocan el alcohol, comprarían estas soluciones a estos vendedores ambulantes y, sin saberlo, serían engañados para consumir soluciones que no sólo contenían alcohol, sino también narcóticos como el opio y/o la cocaína. Con el pretexto de mejorar una absurdamente amplia cornucopia de dolencias, los pacientes desarrollaron una adicción punitiva y/o su salud se vio afectada negativamente por estos primeros "traficantes de drogas".
A medida que el problema crecía, el gobierno federal empezó a tomar nota. Finalmente, el Ley de Alimentos y Drogas Puras fue aprobada en 1906 y condujo a la creación de la Administración de Alimentos y Medicamentos (FDA).
[La FDA tenía una formativas deber de garantizar que los medicamentos lleven declaraciones veraces en el etiquetado y cumplan con ciertos estándares de pureza y concentración.
Recuerde que casi 120 años requisito de etiquetado veraz y la parte de “pureza” de la Ley de Medicamentos y Alimentos Puros de 1906 mientras sigue leyendo sobre las pruebas de verificación de ARNm y la transparencia de los ingredientes.]
¿Qué pruebas de verificación de ingredientes “veraces” y “puros” se están realizando para los productos regulados por la FDA?
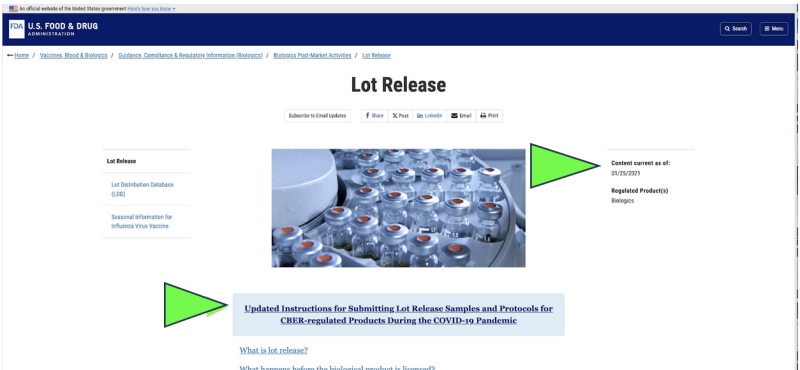
En 2021, la FDA optó por comenzar a monitorear la calidad farmacéutica de Estados Unidos a través de un recogida remota of envío de muestras por correo para medicamentos en lugar de inspecciones de instalaciones en vivo debido a la pandemia de Covid. ¿Era eso legal? ¿Podría alguna vez considerarse eso científicamente apropiado? Hoy, a pesar de que la pandemia ha terminado, la única prueba oficial de liberación de productos farmacéuticos que se realiza actualmente en cualquier Farmacéutica de ARNm de Covid aparece a aun ser realizado por la FDA a través de un proveedor proporcionado por el fabricante, "enviado por correo” muestra según un captura de pantalla del sitio web actual de la FDA. Obviamente, un método de muestreo “enviado por correo” es muy diferente y potencialmente menos confiable que la recolección directa de muestras mediante un método de recolección directa en persona. A pesar de eso, la FDA afirma que tiene “el estándar más alto en todo el mundo para muestreo y pruebas."
Además, la FDA propone seguir avanzando en su política de pruebas remotas "enviadas por correo" con un documento de orientación recientemente propuesto.
Aunque sólo existe como un documento "borrador" de la FDA, los sitios web oficiales de la FDA muestran que El envío de muestras por correo parece ya haberse implementado desde al menos enero de 2021.. La FDA parece estar afirmando los resultados de esas pruebas enviadas por correo como su verificación independiente.
Además, el parte inferior de la primera página del borrador de la FDA El documento propone la expansión de las “pruebas remotas”. Actualmente enumera cada División reguladora de productos de la FDA en la FDA, lo que implica que es una propuesta de política para toda la agencia.
La lista completa incluye:
- Oficina de Asuntos Regulatorios
- Oficina de Política y Respuesta Alimentaria
- Oficina de Productos Combinados
- Centro de Evaluación e Investigación Biológica
- Centro de Evaluación e Investigación de Drogas
- Centro de Dispositivos y Salud Radiológica
- Centro de Seguridad Alimentaria y Nutrición Aplicada
- Centro de Productos de Tabaco
- Centro de Medicina Veterinaria
¿Es apropiado el muestreo de control de calidad “enviado por correo” por la FDA? ¿Qué pasaría si las inspecciones de restaurantes del Departamento de Salud de los estados reflejaran la política de la FDA?
Esta metodología de muestreo "por correo" es igualmente absurda, por ejemplo, que el departamento de salud de un estado monitoree los restaurantes pidiéndoles que "envíen por correo" periódicamente varios artículos de su menú a un centro de pruebas para que los departamentos de salud puedan realizar pruebas de posibles alimentos. contaminación nacida y/o pedir a los restaurantes que se comprometan a probar ellos mismos los elementos del menú. ¿Y si ese restaurante estuviera en China? ¿Y si ese restaurante estuviera en la India? O cualquier otro país conocido por tener una Historia abismal de fraude y control de calidad. ¿problemas?
Esa metodología sería inaceptable tanto para los restaurantes como para las empresas farmacéuticas, por razones que incluyen lo obvio: los fabricantes podrían enviar las muestras que prefieran, no necesariamente muestras por lotes representativas. Obviamente, no es lo mismo que los inspectores de la FDA adquieran muestras durante inspecciones sin previo aviso de toda la instalación.
Bajo la analogía del restaurante, por supuesto todos los restaurantes enviar muestras de calificación "A" lo cual no sería necesariamente representativo de lo que reciben los consumidores.

Control de calidad: ¿Qué son las “pruebas de liberación” farmacéuticas y por qué son importantes?
Hoy en día, la FDA supervisa la calidad y el contenido de $2.7 trillón valor del producto anualmente, pero parece estar suprimiendo las evaluaciones y resultados de verificación de ingredientes críticos. Se supone que la FDA protege a los estadounidenses realizando exhaustivo pruebas analíticas como suma de verificación para garantizar la precisión de los ingredientes. Los resultados de esto deberían ser transparentes para los contribuyentes que financian el Los 6.6 millones de dólares de la FDA presupuesto. Esa verificación científica se denomina farmacéutica”prueba de lanzamiento.” La prueba de lanzamiento es un término técnico que se refiere a un proceso que involucra una variedad de análisis instrumentales utilizados para exhaustivamente productos de prueba para determinar pureza, concentración, consistencia, identidad e impurezas de cualquier tipo.
Toda la FDA nació de ese empleado del “Departamento de Química” de 1862 y de la necesidad de transparencia y verificación de ingredientes. Hoy, ese empleado ha proliferado hasta convertirse en un Todo el departamento de la FDA de 1,300 científicos y personal de apoyo. supuestamente dedicado a la verificación de ingredientes mediante pruebas de liberación de productos farmacéuticos. La FDA Oficina de Calidad Farmacéutica (OPQ) se supone que debe garantizar que los productos farmacéuticos coincidan exactamente con el contenido de los ingredientes enumerados, sin variabilidad de calidad/impureza (cualitativa) o contenido (cualitativa). Las normas que lo exigen son muy específicas y detalladas en 21 CFR § 201.10.
Cómo verifica la FDA las inyecciones de ARNm para el control de calidad:
Los resultados del control de calidad de las pruebas de inyecciones de ARNm fueron particularmente críticos porque son grandes, complejos y se elaboraron rápidamente. Si bien los contribuyentes dependen de la FDA para verificar la calidad de las inyecciones de ARNm y compartir los resultados, la FDA parece obligado a proteger los ingredientes de los fabricantes a expensas incluso de la transparencia más básica con respecto a los productos de ARNm Covid. Si bien la FDA parece estar recolectando muestras, su metodología de “envío por correo” es fundamentalmente defectuosa. Además, la FDA no comparte los resultados de esas pruebas en ningún lugar donde pueda localizarlos.
En otras palabras: durante la pandemia, cuando los estadounidenses recibían a “velocidad vertiginosa” inyecciones de ARNm completamente nuevas y ampliamente implementadas y cuando Estados Unidos dependía más de las obligaciones regulatorias y de calidad de la FDA, la FDA aceptaba “envíos por correo” autoenviados. en” pruebas y/o resultados de control de calidad. ¿La FDA no consideró que Los fabricantes de ARNm admitieron que “luchaban” para responder a la fabricación y estaban “luchando” para mantenerse al día. con los procesos de fabricación? Los fabricantes de ingredientes de ARNm afirmaron además que los esfuerzos para satisfacer las necesidades "no tenían precedentes".
Declaraciones como esta no generan confianza del consumidor en la calidad y son ilustrativas de una enorme mejora de estos productos complejos que deberían justificar especialmente vigilante y el escrutinio en persona de la FDA de las instalaciones y productos manufacturados, pandémicos o no. Un fabricante de ingredientes de ARNm, por ejemplo, afirmó que de repente aumentaron su producción en pliegue 50.
En medio de esa novedosa tecnología impulsada a “velocidad vertiginosa”, ninguno de los 1,300 científicos de OPQ de la FDA exigió inspecciones en vivo, o al menos, se ofreció a hacer algo más que pedir muestras “enviadas por correo” potencialmente cuestionables. ¿para las pruebas?
La pregunta obvia es: ¿Por qué la FDA no recolectó muestras directamente?? Incluso con la pandemia en vigor, la FDA podría haber inspeccionado instalaciones usando trajes protectores o, o al menos muy al menos, optó por recolectar muestras en farmacias, hospitales o en los almacenes de los distribuidores.
Metodología oculta para probar los ingredientes de la inyección de ARNm:
Más allá de la ausencia de resultados de pruebas y resultados de muestras cuestionables “enviados por correo”, la FDA está además ocultando su metodología validada impidiendo que otros realicen sus propios análisis independientes sobre la calidad/pureza de las inyecciones de ARNm.
Analizar de forma independiente la pureza y la posible contaminación de los medicamentos en comparación con la lista de ingredientes es algo que intenté hacer yo mismo cuando conceptualicé el primer producto del mundo. farmacia analítica. Sin embargo, dado que las inyecciones de ARNm son una tecnología novedosa con una lista de ingredientes poco transparente, la metodología de prueba que se necesitaría emplear no es sencilla como lo sería con otros fármacos de molécula pequeña. Cualquiera que intente buscar el almacenamiento, la estabilidad, la especificidad, la química, la sensibilidad o incluso la metodología básica para la validación y/o los resultados de las pruebas se ve bloqueado por un informe de la FDA que contiene redacciones ridículamente invasivas, lo que dificulta incluso la comprensión científica más fundamental sobre cómo evaluar potencialmente resultados o realizar pruebas imposibles.
Como ejemplo visual conmovedor, una sola página redactada en un resumen regulatorio más extenso de la FDA (que se muestra a continuación) es parte de un documento 127 páginas (de las cuales solo se han compartido 63 páginas, y de esas 63 páginas, alrededor del 50% ha sido redactada) sobre cómo evaluar la pureza, concentración y otras medidas analíticas de las inyecciones de ARNm.
aquellos Redacciones de la FDA (b)(4) redacciones detalladas especificadas utilizadas para "proteger secretos comerciales e información comercial o financiera confidencial.” Pero, ¿es realmente apropiado etiquetarlo como “comercial” si la investigación/desarrollo/producto fue financiado con cientos de millones de dólares de los contribuyentes?

Sin una lista de ingredientes o una metodología de prueba, es imposible que alguien fuera de la FDA o los fabricantes sepan con precisión cómo verificar el producto. adulteración (ingredientes alterados o tóxicos) o etiquetado incorrecto (porque una lista completa de ingredientes, incluida la secuencia de nucleótidos y el Configuraciones de nanopartículas lipídicas. son particularmente vagas en la etiqueta del producto).
La falta de metodología es particularmente problemática ya que nuevos datos preliminares que utilizan metodología independiente han mostrado evidencia de Contaminación de ADN en inyecciones de ARNm de Covid.
Entonces, si un individuo externo afirmara haber probado y encontrado una impureza en las inyecciones de ARNm y solicitara a la FDA o a los fabricantes su respuesta, recibiría alguna respuesta que indicaría algo como:
- No utilizó una metodología de prueba validada/apropiada para llegar a sus conclusiones y, por lo tanto, sus análisis no son válidos.
Para eso, el laboratorio independiente intentaría solicitar la metodología de prueba de la documentación aprobada por la FDA (es decir, el documento completo que contiene Figura 5) preguntando: “Está bien, me gustaría probarlo utilizando su metodología aprobada; ¿Nos dirás qué es eso?
- La FDA o el fabricante responderían algo como: “Lo que estamos dispuestos a revelar sobre la metodología empleada que no es confidencial se puede encontrar en línea o mediante una solicitud FOIA de la FDA."... donde se encontrarían el siguiente documento muy redactado, donde cualquier cosa remotamente significativa está cubierta con redacciones (b)(4).
Leyendo entre líneas: es obvio que tanto los fabricantes como la FDA de Estados Unidos no quieren que nadie más que ellos mismos conozca los ingredientes completos o incluso pruebe las inyecciones de ARNm para determinar su pureza y consistencia.
Según funcionarios de la FDA: la fabricación farmacéutica es Altamente Propenso a errores:
Muchos Las cosas pueden salir mal (y de hecho suceden) durante el proceso de fabricación de productos farmacéuticos. Más allá de las posibles inconsistencias con las inyecciones de ARNm/LNP, existen cuestiones cualitativas y cuantitativas que implican cada Producto farmacéutico regulado por la FDA. Incluso la Cámara y el Senado han reconocido formalmente informes sobre el fracaso de la FDA a la hora de asegurar la cadena de suministro farmacéutico de Estados Unidos. La mayoría de los La farmacéutica estadounidense producto de consumidor finalse está produciendo en el extranjero en países como India y China, y otros países con bajos costos laborales están no Bien considerado por sus altos niveles de control de calidad.. El Registro Federal está plagado de informes de Infracciones en plantas de fabricación indias y chinas..
¿La FDA también está certificando estas plantas –incluidas aquellas con un largo historial de violaciones– mediante un sistema de “envío por correo” a la FDA? Escandalosamente, la respuesta a la pregunta es algo que incomodaría mucho a cualquiera preocupado por la calidad farmacéutica.
Mientras que una Six Sigma Aunque el nivel de precisión ha sido durante mucho tiempo el objetivo de la calidad y la seguridad en la fabricación de automóviles, ordenadores, teléfonos móviles y otras manufacturas de alta tecnología, parece haber sido pasado por alto en lo que respecta a la fabricación farmacéutica.
Los funcionarios de la FDA han publicado datos que estiman una imprecisión de 2-3σ (sigma) en la fabricación de productos farmacéuticos. Una cualidad 2σ corresponde a 308,537 defectos por 1,000,000 de oportunidades. (Es probable que haya muchas más de 1,000,000 de oportunidades de error cuando se trata de la fabricación de productos farmacéuticos). La FDA es consciente de esto en los niveles más altos de liderazgo; de hecho, la actual El jefe de la Oficina de Calidad Farmacéutica de la FDA, Michael Kopcha Incluso escribió y publicó el cálculo Six Sigma anterior, lamentando la naturaleza imprecisa de la fabricación farmacéutica. de nuevo en 2017.
La latitud de error para los productos de ARNm y/o sus LNP podría ser incluso menos preciso que el 2-3σ (cuanto menor es el σ, más erróneo es el producto), ya que incluyen material de nucleótidos y LNP novedosos, lo que los hace sustancialmente más complejos que los productos farmacéuticos de molécula pequeña, a pesar de que se desarrollan, fabrican y lanzan en “ velocidad de la luz."
Incluso la FDA y sus funcionarios reconocen una imprecisión inherente en la fabricación, por qué en el amplio mundo del deporte ¿No está cumpliendo la FDA su misión de seguridad al compartir públicamente sus pruebas de liberación de tecnología de ARNm con el público estadounidense que las financia?
¿Antes de 1862 otra vez? ¿Son las inyecciones de ARNm los únicos medicamentos que los estadounidenses no tienen? Solución ¿Información de ingredientes?
La falta de claridad sobre la cantidad de secuencias de inyecciones de ARNm y otra información crítica contrasta directamente con otro medicamento basado en ARN aprobado por la FDA: patisirán (Onpattro®). Onpattro proporciona de forma transparente la secuencia, el peso molecular y la concentración en miligramos de sus productos dentro de la FDA oficial. etiquetado de paquetes como se ilustra en un extracto a continuación:


Falta de especificidad de la dosis de ARNm de Covid: 0.3 ml (o 0.5 ml) de que?
Por el momento, todavía no tenemos información básica sobre los ingredientes de ninguna inyección de ARNm de Covid. Los farmacéuticos sólo saben dar una determinada volumen de fluido, y aparentemente lo hizo sin dudarlo. Normalmente, el etiquetado oficial del paquete de la FDA debe detallar los ingredientes reales en ese volumen, pero no para las etiquetas de ARNm de Covid: simplemente indican 0.3 ml (o 0.5 ml) como "Forma de dosificación y concentración".
Además, como cualquier estudiante de secundaria podría decirle, 0.3/0.5 ml es una volumenno es un fuerza. No conocemos ningún detalle cuantitativo de lo que contiene esos 0.3/0.5 ml, como por ejemplo: ¿Cuántas partículas de LNP? ¿Qué tamaño/morfologías de esos LNP? ¿Cuántas secuencias de ARNm hay en ese volumen?




¿Es esto lo que la FDA considera suficientemente transparente o “etiquetado veraz”?
El extracto de cortar y pegar anterior del prospecto es toda la información que los fabricantes comparten con los consumidores con respecto a la dosis, que es lamentablemente inadecuada en comparación con todas las demás etiquetas de la FDA, o cualquier persona que tenga curiosidad por saber algo más allá de la cantidad de líquido. inyectar y la concentración de 30 o 100 mcg de una secuencia de ARNm no especificada.
La notable imprecisión de esta etiqueta permitida por la FDA parece entrar en conflicto específicamente con su etiqueta de casi 120 años: “Exigir que los alimentos y medicamentos lleven declaraciones veraces en el etiquetado y cumplan con ciertos estándares de pureza y concentración.."
¿Es esto lo que la FDA considera una lista “veraz” de ingredientes? (Ver 21CFR §352y 21 CRF §201.10 con respecto a la “declaración de ingredientes” y “medicamentos y dispositivos mal etiquetados”).
La pregunta es: ¿enumerar ingredientes desconocidos o no específicos que nadie excepto el fabricante puede descifrar? realmente ¿Cumple con el espíritu o los requisitos legales del “etiquetado”? ¿Es esa etiqueta lo que la FDA de Estados Unidos considera “veraz”? ¿De qué lado está la FDA? ¿fabricantes o consumidores?
Además de no especificarse directamente, el número exacto de cadenas de LNP y de ARNm en una inyección de 30 o 100 mcg ni siquiera se puede extrapolar. estequiométricamente o sobre la base de El número de Avogadro, porque la secuencia de ARNm, el peso molecular y/o los componentes/configuraciones de LNP no se proporcionan en ninguna parte del etiquetado oficial de la FDA.
¿Cómo puede alguien saber si la cantidad de hebras de ARNm para codificar la proteína de pico para Covid es proporcional a la carga de inóculo de Covid que uno recibiría de una infección adquirida en la comunidad? Respuesta: ellos no pueden.
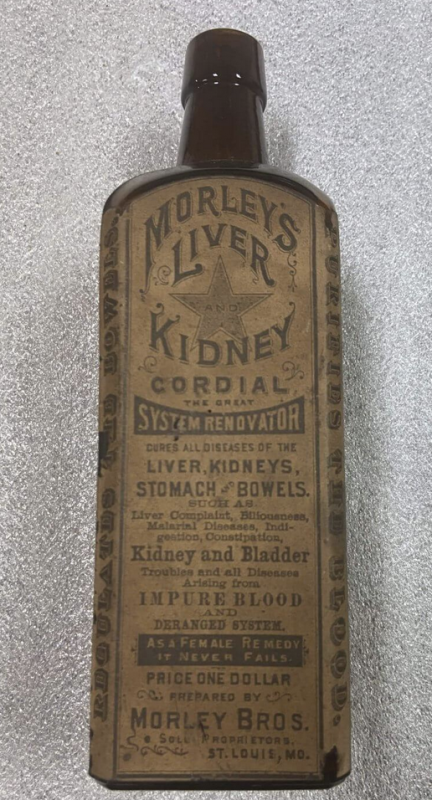
¿Son las inyecciones de ARNm de Covid? Apropiadamente etiquetado/mal etiquetado?
21 CFR 211.125 especifica “Se ejercerá un control estricto sobre el etiquetado emitido para su uso en operaciones de etiquetado de productos farmacéuticos."Pero parece que la FDA fue muy laxa con el etiquetado aprobado de las inyecciones de ARNm de Covid a pesar de que todos los demás medicamentos, incluido Onpattro basado en ARNm, especifican esa información. Históricamente, las decisiones regulatorias de la FDA (como qué información incluir en el etiquetado del producto) se basan en precedentes, y las inyecciones de ARNm de Covid fueron una desviación obvia de los precedentes históricos y legales de la FDA. Esa notable ausencia de datos y falta de claridad se remonta a los días de Cordial de hígado y riñón de Morley a finales del siglo XIX. La diferencia es: en aquel entonces, la FDA no existía, pero hoy hay una FDA con ~1800 empleados, al menos algunos de los cuales aparentemente creían que esta etiqueta era transparente y “veraz”.
Declarar un ingrediente desconocido/indescifrable/oscuro que nadie podría determinar con precisión probablemente no es lo que pretendían los legisladores de la Ley de Alimentos y Medicamentos Puros de 1906 cuando especificaron las reglas de la FDA sobre "etiquetado veraz". Aparte de eso: el hecho de que las dosis se duplican por volumen de diferentes fabricantes (30 mcg/0.3 ml vs 100 mcg/0.5 ml) significa que estas secuencias de ARNm parecen ser muy diferentes en longitud de nucleótidos y, a su vez, tendrían más y diferentes LNP más archivos adjuntos. ¿Eso significa que las secuencias de ARNm utilizadas para transcribir la proteína de pico tienen aproximadamente el doble de tamaño (10 mcg/0.1 ml versus 20 mcg/0.1 ml) en comparación con diferentes fabricantes, o hay algo más que contribuye a la diferencia en la longitud de los nucleótidos?
Para el profano que todavía ha leído hasta este punto (felicitaciones, por cierto): la falta de información detallada en el etiquetado podría ser como anunciar ampliamente una casa en venta, indicando que está hecha de madera y ladrillos, sobre una losa de cemento, pero sin mostrar cualquier imagen de la casa (por ejemplo, secuencia) y no compartir sus pies cuadrados (por ejemplo, peso molecular). En cualquier caso, la falta de información es inadecuada y una desviación de los estándares tradicionales.
Todos los demás medicamentos aprobados por la FDA, incluidos otros medicamentos de ARNm, contienen información completa sobre los ingredientes en sus productos, incluidos una representación estructural y un peso molecular de su producto para que la gente sepa exactamente lo que está adquiriendo.
Es cierto: busca cualquier droga que se te ocurra en el Base de datos de Drugs.com y observe cómo todas las etiquetas proporcionan estructura y/o peso molecular. Prueba de que las inyecciones de ARNm de Covid son una conspicuo excepción a la práctica histórica de aprobación de la FDA y a la regla de “etiqueta veraz”.
Un estudio danés de 2023 detalla una variabilidad clínica significativa entre lotes de inyecciones de ARNm de Covid-19:
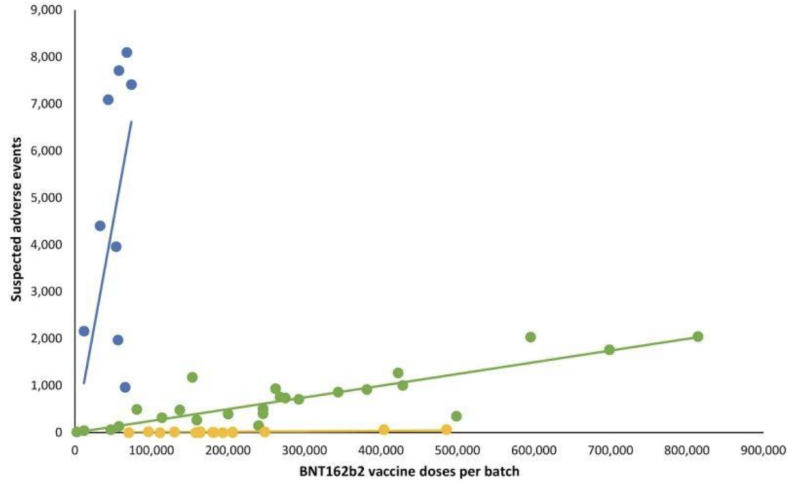
No tener transparencia ni siquiera en la validación de pruebas “enviadas por correo” potencialmente inválidas parece haber dado a los fabricantes un pase a otra parte de importancia crítica de lo que supervisa la FDA: posibles manifestaciones clínicas en variaciones de lotes de inyecciones de ARNm. una retrospectiva Estudio de seguridad danés publicado a principios de 2023 detalla un patrón muy desviado de informes de eventos adversos de las inyecciones de ARNm de BNT162b2 de Pfizer-BioNTech en correlación con el sistema danés de notificación de eventos adversos DKMA.
En el gráfico de líneas que sigue, puntos de diferentes colores representan diferentes lotes de inyecciones de ARNm de Pfizer-BioNTech. Separó lotes en tres categorías diferentes; Número alto-bajo a (casi) ausente de grupos de eventos adversos informados (gráficos azul, verde y amarillo respectivamente).
En otras palabras: Los productos supuestamente “equivalentes” del mismo fabricante parecen tener incidencias muy diferentes de eventos adversos, por lote, y cada uno de esos lotes representa cientos de miles de inyecciones de ARNm.
Cuando se agregaron las líneas de regresión lineal correspondientes, surgió un patrón particular:
Las preguntas importantes sobre la notable disparidad de eventos adversos entre los lotes de ARNm de Covid-19 incluyen:
- ¿Podrían las variaciones de los eventos adversos deberse a variaciones cualitativas o cuantitativas en las secuencias de ARNm o en el número de cadenas de ARNm entre lotes?
- ¿Podrían las variaciones de los eventos adversos deberse a variaciones cualitativas o cuantitativas en el tamaño/morfologías o la cantidad de LNP entre lotes? ¿Qué pruebas se han realizado para Garantizar la seguridad de varios LNP. ¿Se utiliza en inyecciones de ARNm?
- ¿Fueron esos lotes que correspondían con los puntos de datos amarillo versus verde versus azul de alguna manera cualitativa o cuantitativamente diferentes?
- ¿Se vio comprometido el almacenamiento/manipulación posterior a la fabricación en las instalaciones de administración (o en algún otro lugar a lo largo de la cadena de suministro), lo que provocó variabilidad del producto?
- ¿Cuál es la tasa Sigma/error de este y otros productos que se originan en la instalación de fabricación/jefe de turno a cargo de la fabricación en particular?
- ¿Los ingredientes de estos productos de ARNm de Covid procedían de India o China o de otros lugares, según el lote?
- ¿Qué porcentajes de lotes de productos de ARNm de Covid fueron analizados mediante recolección en persona por un inspector de la FDA en comparación con los que se “enviaron por correo” desde el inicio hasta la fecha? ¿Se analizó cada lote utilizando únicamente alguno de estos dos métodos de recolección?
- ¿Realizó la FDA verificación de pruebas de liberación en los lotes del sistema danés de notificación de eventos adversos DKMA? En caso afirmativo, ¿por qué la FDA no publica los resultados de esas pruebas en particular? Si no, ¿por qué no se hicieron las pruebas?
- ¿Existe un problema fundamental con la producción consistente de LNP y/o secuencias de ARNm de manera confiable y sin contaminación?
Los resultados del estudio danés y las preguntas anteriores sobre eventos adversos podrían *comenzar* a abordarse, pero no sin que la FDA comparta de forma independiente los resultados de las pruebas de liberación. Tal como están las cosas, debido a las ubicuas redacciones de la FDA (b)(4), nadie conoce la metodología validada para probar las inyecciones de ARNm de Covid. or exactamente qué lotes en el estudio danés fueron o no analizados or los resultados de esas pruebas por lotes.
…Por otra parte, incluso si la FDA hubiera decidido publicar los resultados de las pruebas por lotes, ¿cómo saben los consumidores si esos resultados son representativos de los lotes especificados, ya que los fabricantes seleccionan por sí mismos qué muestras “enviar por correo”?
No proporcionar transparencia sobre los ingredientes y garantizar la calidad mediante una metodología de muestreo adecuada es un requisito fundamental y básico de la FDA. De hecho, ¡fue la razón principal para la formación de la FDA! ¿No merecen los estadounidenses una mayor transparencia, supervisión y leyes de “etiquetado veraz” en lo que respecta a nuestros productos farmacéuticos, especialmente porque esas leyes se promulgaron hace más de 100 años?
Publicado bajo un Licencia de Creative Commons Atribución Internacional
Para reimpresiones, vuelva a establecer el enlace canónico en el original Instituto Brownstone Artículo y Autor.








