
In primera parte de este artículo, Revisé el marco contractual y regulatorio aplicado por el gobierno de EE. UU. para el desarrollo inicial, la fabricación y la adquisición de las inyecciones de ARNm de Covid, utilizando los acuerdos BioNTech/Pfizer para ilustrar el proceso.
Mostré que la Autorización de Uso de Emergencia (EUA) se otorgó a estos productos en base a ensayos clínicos y procesos de fabricación realizados con
- sin normas jurídicas vinculantes,
- ninguna supervisión o regulación de seguridad proscrita legalmente, y
- No hay compensación legal por parte del fabricante por posibles daños.
En este artículo de seguimiento, proporcionaré un análisis detallado de la documentación subyacente.
Otra autoridad/acuerdo de transacciones (OTA): una vía de adquisición militar
El acuerdo entre el gobierno estadounidense, representado por el Departamento de Defensa (DoD), y Pfizer, en representación de la asociación BioNTech/Pfizer, en julio de 2020, para la compra de una “vacuna para prevenir el COVID-19” no era un contrato de adquisición ordinario.
Fue un acuerdo bajo la Autoridad de Otras Transacciones (OTA), una vía de adquisición que, según Directrices del Departamento de Defensa, se ha utilizado desde 1958 para “permitir que una agencia federal entre en transacciones distintas de contratos, subvenciones o acuerdos de cooperación."
[NEGRITA AÑADIDA]
Se puede encontrar una revisión exhaustiva del uso de OTA por parte del Departamento de Defensa, incluido su historial legal, en el Informe del Servicio de Investigación del Congreso del 22 de febrero de 2019. Este informe, junto con cualquier otro análisis sobre OTA, especifica que es una vía de adquisición alternativa. para fines militares y de defensa. No está destinado, ni se ha utilizado nunca antes de Covid, para nada destinado principalmente a uso civil.
Si buscas Leyes de OTA en el Código de EE. UU., este es el camino que seguirás:
Fuerzas Armadas -> Ley Militar General -> Adquisiciones -> Investigación e Ingeniería -> Acuerdos -> Autoridad del Departamento de Defensa para llevar a cabo ciertos proyectos prototipo
Esta vía legal muestra muy claramente que las leyes OTA están destinadas a la adquisición de prototipos de investigación e ingeniería para las fuerzas armadas.
El Departamento de Defensa tiene autoridad para tres tipos diferentes de OT: (1) OT de investigación, (2) OT prototipo y (3) OT de producción.
Estos tres tipos de OT representan tres etapas de investigación inicial, desarrollo de un prototipo y producción final.
Dentro de esos tres tipos, existen categorías específicas de proyectos a los que la OTA puede postularse:
- Originalmente, según el Descripción general de la OTA proporcionada por el Departamento de Defensa, la Autoridad para Otras Transacciones estaba "limitada a aplicarse a armas o sistemas de armas propuestos para ser adquiridos o desarrollados por el Departamento de Defensa".
- Posteriormente, la OTA se amplió para incluir "cualquier proyecto prototipo directamente relacionado con la mejora de la eficacia de la misión del personal militar y las plataformas, sistemas, componentes o materiales de apoyo propuestos para ser adquiridos o desarrollados por el Departamento de Defensa, o con la mejora de plataformas, sistemas, componentes". , o materiales en uso por las Fuerzas Armadas”.
Hasta ahora, nada de eso parece una vía de adquisición para millones de nuevos productos médicos destinados principalmente al uso civil.
¿Existe alguna excepción para el uso civil de OTA que pueda aplicarse a las vacunas de ARNm de Covid?
El Ley de Autorización de Defensa Nacional para el año fiscal 2004 (P.L. 108-136) contenía una sección que otorgaba autoridad para otras transacciones al “jefe de una agencia ejecutiva que se dedica a investigación básica, investigación aplicada, investigación avanzada y proyectos de desarrollo” que “tienen el potencial de facilitar la defensa contra el terrorismo o la recuperación contra el terrorismo o el terrorismo nuclear, biológico o nuclear”. ataque químico o radiológico”.
Esta disposición se prorrogó hasta 2018, pero no parece haberse extendido más allá de ese año. Además, tenga en cuenta que incluso en este caso excepcional de uso de OTA por parte del Departamento de Defensa, la situación debe involucrar terrorismo o un ataque con armas de destrucción masiva (QBRN).
¿Qué otras leyes de OTA podrían aplicarse?
El informe CRS de 2019 citado anteriormente proporciona este cuadro, que muestra que algunas agencias que no pertenecen al Departamento de Defensa tienen alguna OTA o autoridades relacionadas:
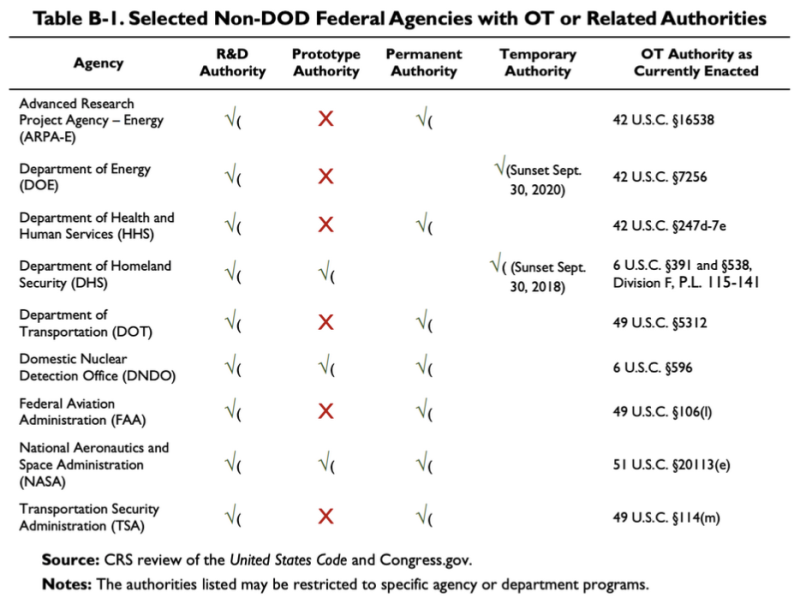
Según esta tabla, el Departamento de Salud y Servicios Humanos (HHS) tiene algunas autoridades de investigación y desarrollo (I+D) y otras transacciones. La ley relativa a la La autoridad OT del HHS es 42 U.S.C. §247d-7e.
¿Dónde se aloja esta ley y qué dice?
Salud y Bienestar Público -> Servicio de Salud Pública -> Poderes y Deberes Generales -> Cooperación Federal-Estatal -> Autoridad de Investigación y Desarrollo Biomédico Avanzado (BARDA) -> Autoridades de Transacciones
Por lo tanto, hay un lugar en la ley relacionada con la salud y el bienestar de los civiles donde la OTA podría ser aplicable, aunque es válida. sólo para investigación y desarrollo, no para prototipos o fabricación.
La ley establece que el secretario de BARDA tiene Autoridad OT
con respecto a un producto que es o puede convertirse en un contramedida calificada o un producto calificado de pandemia o epidemia, actividades que predominantemente—
(i) se llevan a cabo después de la investigación básica y el desarrollo preclínico del producto; y
(ii) estén relacionados con la fabricación del producto a escala comercial y en una forma que satisfaga los requisitos reglamentarios según la Ley Federal Ley de Alimentos, Drogas y Cosméticos [21 USC 301 et seq.] o bajo artículo 262 de este título.
[NEGRITA AÑADIDA]
Los “requisitos regulatorios” enumerados en la ley significan que sería imposible para BARDA/HHS celebrar acuerdos –incluso solo de I+D– para cualquier producto médico (como las vacunas de ARNm) que no se haya sometido a rigurosas pruebas de seguridad y una estricta supervisión de fabricación.
La “asociación” del HHS con el Departamento de Defensa eludió las leyes de protección civil
Para resumir la situación de otras autoridades/acuerdos de transacciones con respecto a las autoridades civiles, en general, y las vacunas de ARNm de Covid, en particular:
- La OTA fue escrita y codificada como una forma para que los militares adquirieran armas y otros sistemas y equipos necesarios sin mucha burocracia. Abarca investigación y desarrollo, prototipos y posterior fabricación.
- La única OTA para una agencia de salud pública es la del HHS y solo cubre Investigación y Desarrollo, no prototipos ni fabricación.
- Incluso la OTA de I+D otorgada al HHS sigue exigiendo que los productos se fabriquen “en una forma que satisfaga los requisitos reglamentarios” de seguridad de medicamentos y vacunas.
En otras palabras: no hay manera de que el HHS hubiera podido utilizar su muy limitada OTA para firmar contratos por cientos de millones de nuevos productos médicos.
Entonces, ¿qué hizo el HHS?
Como señaló la Oficina de Responsabilidad Gubernamental (GAO) en su Informe de julio de 2021 sobre “Contratación de Covid-19”: El HHS "se asoció" con el Departamento de Defensa para "aprovechar las autoridades de OTA del Departamento de Defensa... de las que carecía el HHS". (P. 24)
¿Cuáles son las autoridades de OT del Departamento de Defensa para productos médicos?
Como se mencionó, la OTA tiene como objetivo ayudar a los militares a obtener equipos y tecnología sin muchos problemas burocráticos. Ninguna de las leyes originales relativas a la OTA mencionaba nada más que "plataformas, sistemas, componentes o materiales" destinados a "mejorar la eficacia de la misión del personal militar".
Pero cinco años antes del Covid, se introdujo un uso excepcional de la OTA:
En 2015, Departamento de Defensa anunció el establecimiento del Consorcio de Contramedidas Médicas QBRN, cuyo propósito era utilizar la vía de adquisición de OTA para “trabajar con el Departamento de Defensa para desarrollar contramedidas médicas químicas, biológicas, radiológicas y nucleares autorizadas por la FDA”. [FDA = Administración de Alimentos y Medicamentos]
Como se describe en el anuncio de 2015, esto incluía “tecnologías prototipo para contramedidas médicas terapéuticas dirigidas a objetivos de toxinas virales, bacterianas y biológicas de interés para el Departamento de Defensa”. La lista de agentes incluía los principales patógenos de guerra biológica, como el ántrax, el ébola y el marburgo.
El anuncio continuó especificando que “las tecnologías habilitadoras pueden incluir modelos animales de enfermedades y patogénesis virales, bacterianas o de toxinas biológicas (múltiples rutas de exposición), ensayos, tecnologías de diagnóstico u otras tecnologías de plataforma que puedan aplicarse al desarrollo de MCM aprobados o autorizados. [contramedidas médicas]”.
Aunque esto todavía no se parece en nada a la producción de 100 millones de nuevas vacunas para uso civil, sí proporciona más margen de maniobra a la OTA que la muy limitada Autoridad para Otras Transacciones otorgada al HHS.
Si bien la OTA del HHS requiere el cumplimiento de extensas regulaciones de desarrollo y fabricación, la vía de la OTA para que el Departamento de Defensa desarrolle contramedidas médicas solo requiere una “licencia de la FDA”.
Por lo tanto, utilizando otras autoridades de transacciones del Departamento de Defensa, en teoría sería posible eludir cualquier norma de seguridad, dependiendo de los requisitos para la concesión de licencias de la FDA de un producto generado por OTA. Como veremos, en el caso de las vacunas de ARNm de Covid, se otorgó una Autorización de Uso de Emergencia, sin requerir ningún tipo de supervisión legal de seguridad.
Autorización de uso de emergencia (EUA)
Así es como la Administración de Alimentos y Medicamentos (FDA) describe sus poderes de EUA:
Sección 564 de la Ley FD&C (21 USC 360bbb – 3) permite a la FDA fortalecer la protección de la salud pública contra agentes biológicos, químicos, nucleares y radiológicos.
Con esta autoridad de EUA, la FDA puede ayudar a garantizar que se puedan utilizar contramedidas médicas en emergencias para diagnosticar, tratar o prevenir enfermedades o afecciones graves o potencialmente mortales causadas por agentes biológicos, químicos, nucleares o radiológicos cuando no existen medidas adecuadas y aprobadas. y alternativas disponibles (entre otros criterios).
Es extremadamente importante entender que estos poderes de EUA se otorgaron en 2004 bajo circunstancias muy específicas relacionadas con la preparación para ataques con armas de destrucción masiva, también conocidas como agentes QBRN (químicos, biológicos, radiológicos y nucleares).
Como se explica en la Ley de Salud de Harvard Law,
En última instancia, fue la Guerra contra el Terrorismo la que daría lugar a la autorización de uso de emergencia. Después de los acontecimientos del 11 de septiembre de 2001 y los posteriores ataques por correo con ántrax, el Congreso promulgó la Ley del Proyecto Bioshield de 2004. La ley pedía miles de millones de dólares en asignaciones para la compra de vacunas en preparación para un ataque bioterrorista y para el almacenamiento de contramedidas de emergencia. Para poder actuar rápidamente en caso de emergencia, el Congreso permitió a la FDA autorizar productos formalmente no aprobados para uso de emergencia contra una amenaza a la salud y seguridad públicas (sujeto a una declaración de emergencia por parte del HHS). El grabar indica que el Congreso se centró específicamente en la amenaza del bioterrorismo, no en prepararse para una pandemia de origen natural.
El redacción de la ley EUA subraya el hecho de que estaba destinado a ser utilizado en situaciones en las que intervienen armas de destrucción masiva. Estas son las 4 situaciones en las que se puede emitir EUA:
- una determinación del Secretario de Seguridad Nacional de que existe una emergencia interna, o un potencial significativo para una emergencia interna, que implica un mayor riesgo de ataque con un agente o agentes biológicos, químicos, radiológicos o nucleares;
- una determinación del Secretario de Defensa de que existe una emergencia militar, o un potencial significativo para una emergencia militar, que implica un mayor riesgo para las Naciones Unidas. Zonas fuerzas militares, incluido el personal que opera bajo la autoridad del Título 10 o el Título 50, de ataque con:
- un agente o agentes biológicos, químicos, radiológicos o nucleares; o
- un agente o agentes que puedan causar, o estén asociados de otro modo con, un riesgo específico y que ponga en peligro la vida de United Zonas fuerzas militares;
- una determinación por parte del Secretario que existe una emergencia de salud pública, o un potencial significativo para una emergencia de salud pública, que afecta, o tiene un potencial significativo de afectar, la seguridad nacional o la salud y seguridad de las Naciones Unidas. Zonas ciudadanos residentes en el extranjero, y que involucre un agente o agentes biológicos, químicos, radiológicos o nucleares, o una enfermedad o condición que pueda ser atribuible a dicho agente o agentes; o
- la identificación de una amenaza importante de conformidad con la sección 319F-2 de la Ley de servicios de salud pública [42 USC 247d-6b] suficiente para afectar la seguridad nacional o la salud y seguridad de las Naciones Unidas. Zonas ciudadanos residentes en el extranjero.
En ninguna parte de estas cuatro situaciones se menciona una epidemia, pandemia o cualquier otro tipo de situación de salud pública que ocurra naturalmente y que no sea causada por “agentes biológicos, químicos, radiológicos o nucleares”.
¿Podría el SARS-CoV-2 calificar como tal agente?
Si buscas la definición de “agentes biologicos”en el Código Legal de EE. UU., seguirá el siguiente camino:
Delitos y Procedimiento Penal -> Delitos -> Armas Biológicas -> Definiciones
Así, en el contexto de la legislación estadounidense, el término “agentes biológicos” significa armas biológicas, y el uso de tales agentes/armas se considera un delito.
Wikipedia proporciona esto definición:
Un agente biológico (también llamado bioagente, agente de amenaza biológica, agente de guerra biológica, arma biológica o arma biológica) es un bacteria, virus, protozoario, parásito, hongo, o toxina que puede usarse intencionalmente como arma en bioterrorismo or guerra biológica (BN).
¿Sobre qué base jurídica se emitió la EUA para las vacunas de ARNm de Covid?
Parecería, según las leyes relativas a EUA, que ninguna de las cuatro situaciones posibles descritas en la ley podría aplicarse a un producto destinado a prevenir o tratar una enfermedad causada por un patógeno natural.
Sin embargo, esta ley se utilizó para autorizar las vacunas de ARNm contra el Covid.
Dadas las cuatro opciones enumeradas en la ley EUA, la que se utilizó para las “contramedidas” de Covid fue
C) una determinación por parte del Secretario que existe una emergencia de salud pública, o un potencial significativo para una emergencia de salud pública, que afecta, o tiene un potencial significativo de afectar, la seguridad nacional o la salud y seguridad de las Naciones Unidas. Zonas ciudadanos que viven en el extranjero, y que involucre un agente o agentes biológicos, químicos, radiológicos o nucleares, o una enfermedad o condición que pueda ser atribuible a dicho agente o agentes.
Cuándo aplicado específicamente a Covid, así quedó redactado:
el Secretario del Departamento de Salud y Servicios Humanos (HHS) determinó que existe una emergencia de salud pública que tiene un potencial significativo para afectar la seguridad nacional o la salud y seguridad de los ciudadanos estadounidenses que viven en el extranjero, y que involucra el virus que causa el Coronavirus. Enfermedad 2019 (COVID-19)…
No hay duda aquí de que “el virus que causa el COVID-19” se considera equivalente a “un agente o agentes biológicos, químicos, radiológicos o nucleares”.
También es importante tener en cuenta que la “determinación de emergencia de salud pública” de la EUA es completamente independiente y no depende de ninguna manera de otras declaraciones de emergencia de salud pública, como las que hicieron la OMS, el gobierno de EE. UU. , y el Presidente al inicio de la pandemia de Covid-19.
Entonces, incluso cuando la OMS, el gobierno de los EE. UU. y el Presidente declaren que la pandemia ha terminado, todavía puede haber una Autorización de Uso de Emergencia si el Secretario del HHS continúa afirmando que existe la situación descrita en la sección C).
En cuanto a todas las EUA para cientos de productos médicos relacionados con Covid, es muy difícil ver cómo el secretario del HHS podría justificar la afirmación de que “existe una emergencia de salud pública que tiene un potencial significativo para afectar la seguridad nacional o la salud y seguridad de los ciudadanos estadounidenses que viven en el extranjero” en la mayoría, si no en todos, de estos casos.
“Criterios estatutarios” adicionales para que la FDA otorgue la autorización de uso de emergencia
Una vez que el Secretario del HHS declara que existe una emergencia de salud pública que justifica una EUA, basándose en una de las cuatro situaciones enumeradas en la ley, hay cuatro “criterios estatutarios” más que deben cumplirse para que la FDA emita la EUA. . Así es como la FDA explica estos requisitos:
- Enfermedad o afección grave o potencialmente mortal
Para que la FDA emita una EUA, los agentes QBRN mencionados en la declaración EUA del Secretario del HHS deben ser capaces de causar una enfermedad o afección grave o potencialmente mortal.
NOTA: Este criterio repite la especificación de un agente QBRN, que se define legalmente como un arma utilizada para cometer un delito.
- Evidencia de efectividad
Los productos médicos que pueden considerarse para una EUA son aquellos que “pueden ser efectivos” para prevenir, diagnosticar o tratar enfermedades o afecciones graves o potencialmente mortales que pueden ser causadas por uno o varios agentes QBRN identificados en la declaración de autorización del Secretario del HHS. emergencia o amenaza de emergencia bajo la sección 564(b).
El estándar de “puede ser eficaz” para las EUA proporciona un nivel de evidencia más bajo que el estándar de “eficacia” que utiliza la FDA para la aprobación de productos. La FDA tiene la intención de evaluar la eficacia potencial de un posible producto EUA caso por caso mediante un análisis de riesgo-beneficio, como se explica a continuación.
[NEGRITA AÑADIDA]
PREGUNTA LEGAL: ¿Cómo puede alguien afirmar legalmente que un producto autorizado bajo EUA es “seguro y efectivo” si el estándar legal para EUA es “puede ser efectivo” y la FDA declara que este es un “nivel de evidencia más bajo” que el estándar utilizado? para aprobaciones periódicas de productos?
- Análisis de riesgo / beneficio
Un producto puede ser considerado para una EUA si el Comisionado determina que los beneficios conocidos y potenciales del producto, cuando se usa para diagnosticar, prevenir o tratar la enfermedad o condición identificada, superan los riesgos conocidos y potenciales del producto.
Para determinar si los beneficios conocidos y potenciales del producto superan los riesgos conocidos y potenciales, la FDA tiene la intención de mirar la totalidad de la evidencia científica para realizar una determinación global riesgo-beneficio. Tal evidencia, que podría surgir de una variedad de fuentes, puede incluir (pero no se limita a): resultados de ensayos clínicos nacionales y extranjeros, datos de eficacia in vivo de modelos animales y datos in vitro, disponible para consideración de la FDA. La FDA también evaluará la calidad y cantidad del evidencia disponible, dado el estado actual del conocimiento científico.
[NEGRITA AÑADIDA]
NOTA LEGAL: No existe un estándar legal ni definiciones legales de lo que significa que los “beneficios conocidos y potenciales” superen los “riesgos conocidos y potenciales”. Tampoco existe una definición jurídica cualitativa o cuantitativa de lo que constituye una “evidencia disponible” aceptable en la que “puede basarse” el análisis riesgo-beneficio. Podría no haber ninguna evidencia real, pero sí la creencia de que un producto tiene muchos beneficios potenciales y no muchos riesgos potenciales, y eso satisfaría este "requisito legal".
- Sin alternativas
Para que la FDA emita una EUA, no debe haber ninguna alternativa adecuada, aprobada y disponible al producto candidato para diagnosticar, prevenir o tratar la enfermedad o afección. Un posible producto alternativo puede considerarse “no disponible” si no hay suficientes suministros de la alternativa aprobada para satisfacer plenamente la necesidad de emergencia.
CONSULTA LEGAL: Aparte de la atroz y potencialmente criminal difamación/prohibición de tratamientos alternativos para el Covid-19 como la ivermectina y la hidroxicloroquina, ¿en qué momento hubo una alternativa aprobada para “prevenir el Covid-19” (la única función para la que se compraron las vacunas de ARNm)? ) – Paxlovid, por ejemplo, ¿lo que haría que una EUA para las vacunas de ARNm ya no fuera legal?
Así es como se cumplieron todos estos "criterios legales" en la práctica real Autorización de uso de emergencia para las vacunas de ARNm de BioNTEch/Pfizer Covid:
He concluido que el uso de emergencia de la vacuna Pfizer-BioNTech COVID-19 para la prevención de COVID-19 cuando se administra como se describe en el Alcance de la autorización (Sección II) cumple con los criterios para la emisión de una autorización según la Sección 564(c) de la Ley, porque:
- El SARS-CoV-2 puede causar una enfermedad o afección grave o potencialmente mortal, incluida una enfermedad respiratoria grave, a los seres humanos infectados por este virus;
- Según la totalidad de la evidencia científica disponible para la FDA, es razonable creer que la vacuna Pfizer-BioNTech COVID-19 puede ser eficaz para prevenir el COVID-19, y que, cuando se utiliza en las condiciones descritas en esta autorización, los beneficios conocidos y potenciales de la vacuna Pfizer-BioNTech COVID-19 cuando se usa para prevenir COVID-19 superar sus riesgos conocidos y potenciales; y
- No existe una alternativa adecuada, aprobada y disponible al uso de emergencia de la vacuna COVID-19 de Pfizer-BioNTech para prevenir COVID-19.
[NEGRITA AÑADIDA]
NOTA: El único contexto en el que la FDA sopesó los posibles beneficios y riesgos de la vacuna, y en el que determinó que “podría ser efectiva” fue en la prevención del Covid-19.
No hay consideración, no hay evidencia de beneficio real o potencial, y no se determina que exista alguna efectividad potencial para que la vacuna haga cualquier otra cosa, incluyendo: reducir el riesgo de enfermedad grave, reducir el riesgo de hospitalización, reducir el riesgo de muerte. , reduciendo el riesgo de cualquier condición real o potencialmente relacionada con Covid-19.
POR LO TANTO, uno podría cuestionar razonablemente la legalidad de cualquier afirmación de que la vacuna es “segura y eficaz” en el contexto de cualquier otra cosa que no sea “cuando se usa para prevenir el COVID-19”, algo que se sabía que las vacunas NO HACEN muy poco después de su lanzamiento. introducido.
Si a la gente se le dijera que las vacunas de ARNm de BioNTech/Pfizer son “seguras y efectivas” para cualquier otra cosa que no sea prevenir el Covid-19, y si se les amenazara con cualquier consecuencia por no tomar la vacuna para algo más que prevenir el Covid-19, ¿podrían hacerlo? ¿Tiene un argumento legítimo de que fueron obligados ilegalmente a tomar un producto no aprobado bajo reclamos fraudulentos?
Requisitos de tercer nivel para EUA para productos no aprobados
Una vez que tengamos la declaración de emergencia específica de la EUA, y una vez que la FDA declare que el producto puede ser efectivo y que cualquier evidencia disponible (de cero a infinito) demuestre que sus beneficios superan sus riesgos (según lo determinado por lo que la FDA cree que podrían ser), hay una capa más de regulación no relacionada con la seguridad ni la eficacia.
Así es como un Informe del Servicio de Investigación del Congreso de 2018 sobre EUA explica esto:
FFDCA §564 ordena a la FDA que imponga ciertas condiciones requeridas en una EUA y permite condiciones discrecionales adicionales cuando corresponda. Las condiciones requeridas varían dependiendo de si la EUA es para un producto no aprobado o para un uso no aprobado de un producto aprobado. Para un producto no aprobado, las condiciones de uso deben:
(1) garantizar que los profesionales de atención médica que administran el producto reciban la información requerida;
(2) garantizar que las personas a quienes se administra el producto reciban la información requerida;
(3) prever el seguimiento y notificación de eventos adversos asociados con el producto; y
(4) prever el mantenimiento de registros y la presentación de informes por parte del fabricante.
PREGUNTA LEGAL: ¿Qué es exactamente la “información requerida”? Sabemos que se informó a las personas que las vacunas recibieron una Autorización de uso de emergencia. Pero ¿les dijeron que esto significa “un nivel de evidencia más bajo” que el requerido para las afirmaciones de “seguridad y eficacia” sobre otros productos médicos? ¿Se les informó que existen diferentes niveles de “seguro y efectivo” dependiendo de si un producto tiene EUA u otro tipo de autorización?
NOTA: La ley exige que haya una manera de monitorear y reportar eventos adversos. Sin embargo, no indica quién monitorea, cuáles son los estándares para la presentación de informes y cuál es el umbral para tomar medidas basadas en los informes.
EUA en comparación con cualquier otra vía de aprobación de medicamentos/vacunas
Como investigador/escritor Sasha Latypova Como ha señalado, muchas personas se sintieron confundidas con EUA, porque se parece mucho a EAU, que significa "Uso de acceso ampliado". Este es un tipo de autorización otorgada a productos médicos cuando existe una necesidad urgente por parte de un grupo particular de pacientes (por ejemplo, pacientes con cáncer en etapa IV cuya esperanza de vida se mide en meses) que están dispuestos a arriesgarse a sufrir eventos adversos e incluso la muerte a cambio de acceso. a un tratamiento experimental.
La Autorización de uso de emergencia no está relacionada de ninguna manera con el Uso de acceso ampliado ni tiene ningún parecido con él.
Las diversas vías legales para autorizar productos médicos se presentan claramente en una tabla destacada por un investigador jurídico. katherine vatio. La tabla es parte de una presentación de 2020 para una sesión de aprendizaje conjunta de la FDA y los CDC: Actualizaciones regulatorias sobre el uso de contramedidas médicas.
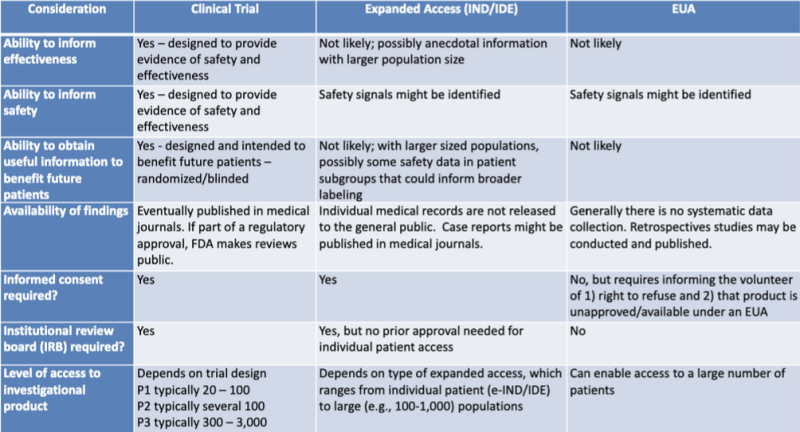
Esta tabla muestra muy claramente que es poco probable que el proceso de EUA proporcione información sobre la efectividad del producto, no está diseñado para proporcionar evidencia de seguridad, no es probable que proporcione información útil para beneficiar a futuros pacientes, no implica una recopilación sistemática de datos, no requiere estudios retrospectivos, sin consentimiento informado y sin junta de revisión institucional.
Además, en un 2009 Instituto de Medicina de la publicación Académica Nacional, también destacado por Watt, titulado “Contramedidas médicas: dispensación de autorizaciones de uso de emergencia y el modelo postal – Resumen del taller” encontramos esta declaración en la p. 28:
Es importante reconocer que una EUA no es parte del camino del desarrollo; es una entidad completamente separada que se utiliza sólo durante situaciones de emergencia y no forma parte del proceso de aprobación de medicamentos.
¿Significa esto que las aprobaciones de contramedidas Covid-19 basadas en EUA eran ilegales? ¿Significa esto que no existe una forma legal de afirmar que un producto EUA es “seguro y eficaz” porque NO ES PARTE DEL PROCESO DE APROBACIÓN DEL MEDICAMENTO?
Conclusión
Es eminentemente evidente, dada toda la información contenida en este artículo y en el anterior. Parte 1, que las vacunas de ARNm de BioNTach/Pfizer Covid fueron desarrolladas, fabricadas y autorizadas conforme a leyes militares reservadas para situaciones de emergencia que involucran guerra biológica/terrorismo, no enfermedades naturales que afecten a toda la población civil.
Por lo tanto, el cumplimiento de las regulaciones y la supervisión que esperamos encontrar cuando un producto se considera “seguro y eficaz” para toda la población civil no era un requisito legal.
¿Se puede utilizar este análisis para cuestionar la legalidad de la afirmación “segura y eficaz” de aquellos funcionarios gubernamentales que sabían lo que implicaba la EUA? ¿Existen otras ramificaciones legales?
Espero que sí.
Es importante destacar que en las impugnaciones legales a las vacunas de ARNm de Covid presentadas hasta ahora, no ha habido fallos (que yo sepa) sobre si la ley militar, como la OTA y la EUA, puede aplicarse a situaciones civiles. Sin embargo, ha habido una declaración del juez del Tribunal de Distrito Michael Truncale, en su sobreseimiento del caso del denunciante Brook Jackson contra Ventavia y Pfizer, eso es importante tenerlo en cuenta.
Aquí el juez reconoce que el acuerdo para las vacunas de ARNm de BioNTech/Pfizer era una OTA militar, pero se niega a pronunciarse sobre su aplicabilidad a las circunstancias no militares (enfermedades naturales, 100 millones de dosis en su mayoría no para uso militar) bajo las cuales se emitió:
El hecho de que tanto el personal militar como los civiles recibieran la vacuna no indica que adquirirla fuera irrelevante para mejorar la eficacia de la misión militar. Más importante aún, la Sra. Jackson está pidiendo de hecho a este Tribunal que anule la decisión del Departamento de Defensa de ejercer la autoridad para otras transacciones para comprar la vacuna de Pfizer. Pero como la Corte Suprema de los Estados Unidos ha enfatizado durante mucho tiempo, las “decisiones complejas, sutiles y profesionales en cuanto a la composición, entrenamiento, equipamiento y control de una fuerza militar son juicios militares esencialmente profesionales”. Gilligan contra Morgan, 413 U.S. 1, 10 (1973). Por tanto, es “difícil concebir un área de actividad gubernamental en la que los tribunales tengan menos competencia”. Identificación. Este Tribunal no vetará las sentencias del Departamento de Defensa relativas a la eficacia de la misión durante una emergencia nacional.
Este es solo uno de los muchos obstáculos legales que quedan en la batalla para prohibir en última instancia todos los productos de ARNm aprobados durante la emergencia de Covid-19 y cualquier producto de ARNm posterior cuya aprobación se haya basado en el proceso de aprobación de Covid-19.
Publicado bajo un Licencia de Creative Commons Atribución Internacional
Para reimpresiones, vuelva a establecer el enlace canónico en el original Instituto Brownstone Artículo y Autor.








